
Nov 16, 2016 4:31:00 PM
Gene expression is tightly regulated and controls the development and maintenance of cells within all organisms. Misregulation of genes can cause adverse effects, for example in humans this could contribute to the initiation and progression of a disease. A technology which is used to identify these misregulated genes is ChIP-sequencing, also known as ChIP-seq.
ChIP-seq is a powerful method that provides the genomic location of regulatory regions (for example transcription-factor binding and histone modifications) in living cells. ChIP-seq combines chromatin immunoprecipitation (ChIP) with massively parallel DNA sequencing to identify the binding sites of DNA-associated proteins. The sites of enrichment are identified using an antibody against the protein of interest to crosslink DNA-protein complexes.
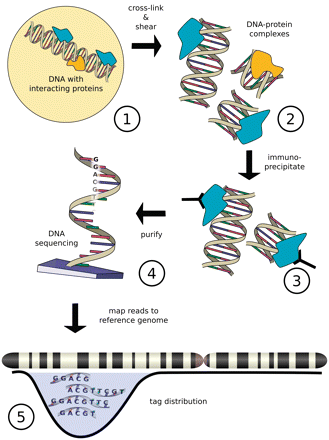 [Image from Szalkowski, A.M, and Schmid, C.D.(2010) http://ccg.vital-it.ch/chipseq/doc/chipseq_tutorial_intro.php]
[Image from Szalkowski, A.M, and Schmid, C.D.(2010) http://ccg.vital-it.ch/chipseq/doc/chipseq_tutorial_intro.php]
The sensitivity of this technology depends on the
-
depth of the sequencing run (i.e. the number of mapped sequence tags)
-
size of the genome
-
specificity of the antibody
For more details, try reading this paper.
As with many high-throughput sequencing approaches, genome wide ChIP-seq generates extremely large data sets (in the order of 30–100 million mapped reads). Appropriate computational analysis methods are required to identify the uniquely mapped reads, the minimal signal strength for a mammalian sample is suggested to be 20 million.
To predict DNA-binding sites from these sequences reads, differential peak calling methods have been developed that identify significant differences in ChIP-seq signals from distinct biological conditions. Eagle Genomics have worked with experts to develop proficiency with the SeqMonk tool. SeqMonk allows visualisation and analysis of any mapped sequence data (BAM/SAM etc) against an annotated genome. Quantitation and statistical analysis of data can be performed to find the regulatory regions of interest and allow comparisons of these regions between data sets. The image below shows enrichment of reads (shown by the height of the bars) from four samples that map to a gene of interest - we can see differential enrichment of reads across a gene between the samples.
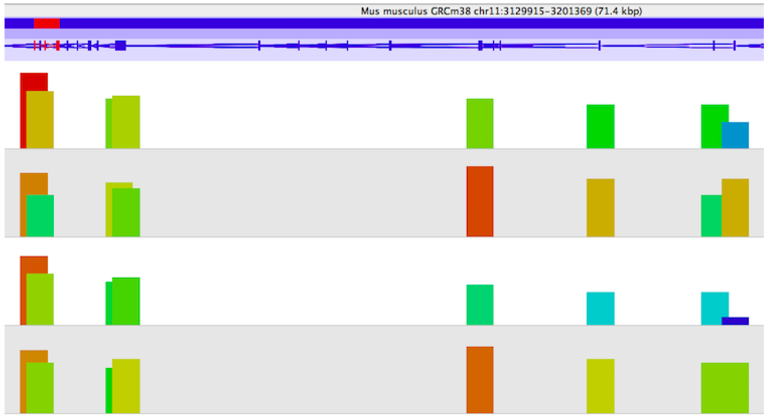
ChIP-seq has become a valuable and widely used approach for mapping the genomic location of transcription-factor binding and histone modifications in living cells. Analysis of these large datasets to determine the regulatory regions that are important for your condition of interest is a complicated process. There are many tools are available, though at Eagle Genomics we have applied our expertise to SeqMonk for quantitation analysis. It is obviously very important to get reliable results, as this is essential to fully understand biological processes and disease states, and helping our customers with this has been a rewarding process.